In collaboration with Dr. Steven Weinman, M.D., Ph.D. (Director of the Liver Center at University of Kansas, Kansas City, Kansas), Cardax evaluated the capacity for a proprietary astaxanthin (ASTX) prodrug (CDX-085) to ameliorate oxidative stress-induced hepatic dysfunction in an animal model of disease.
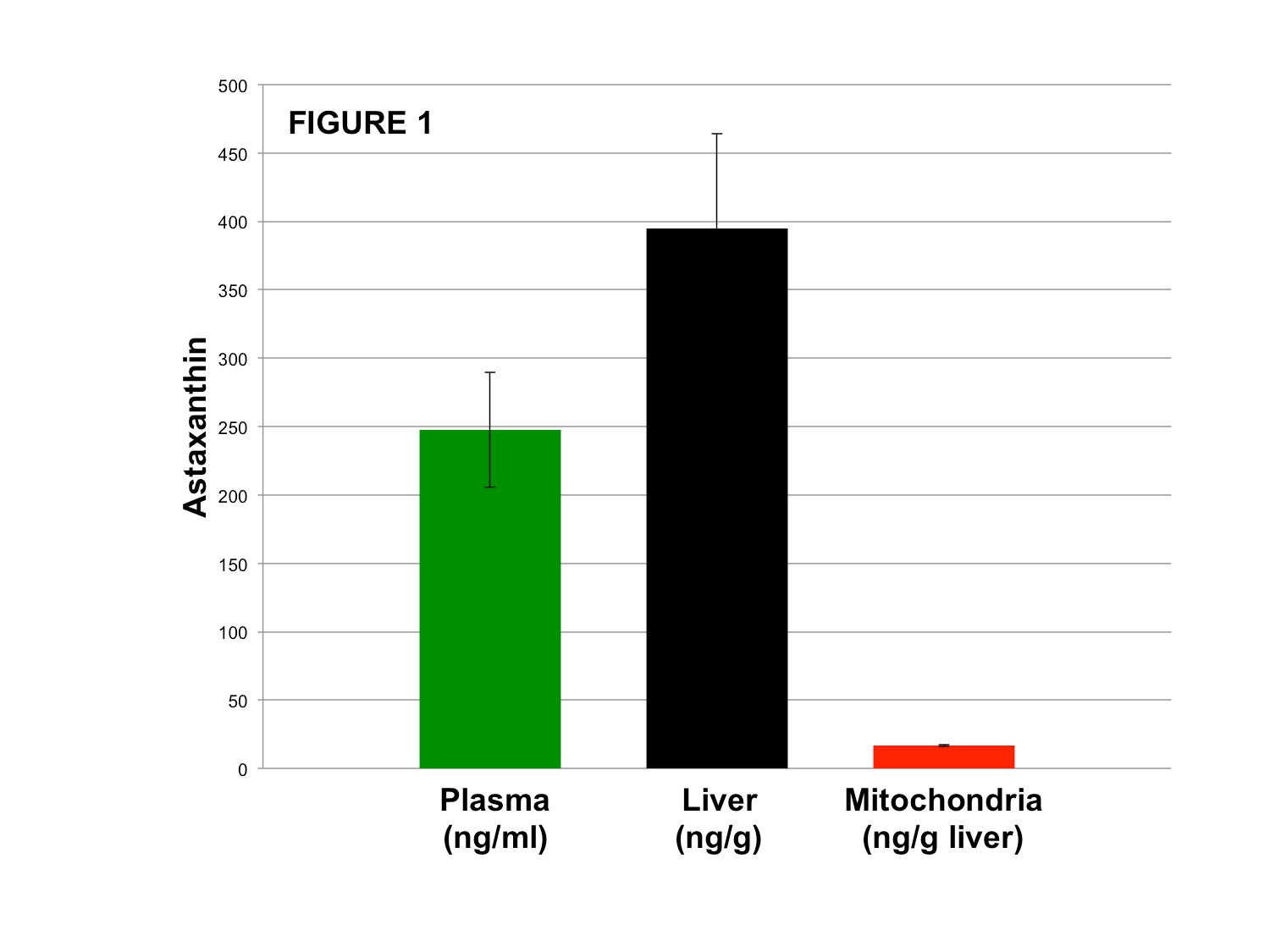
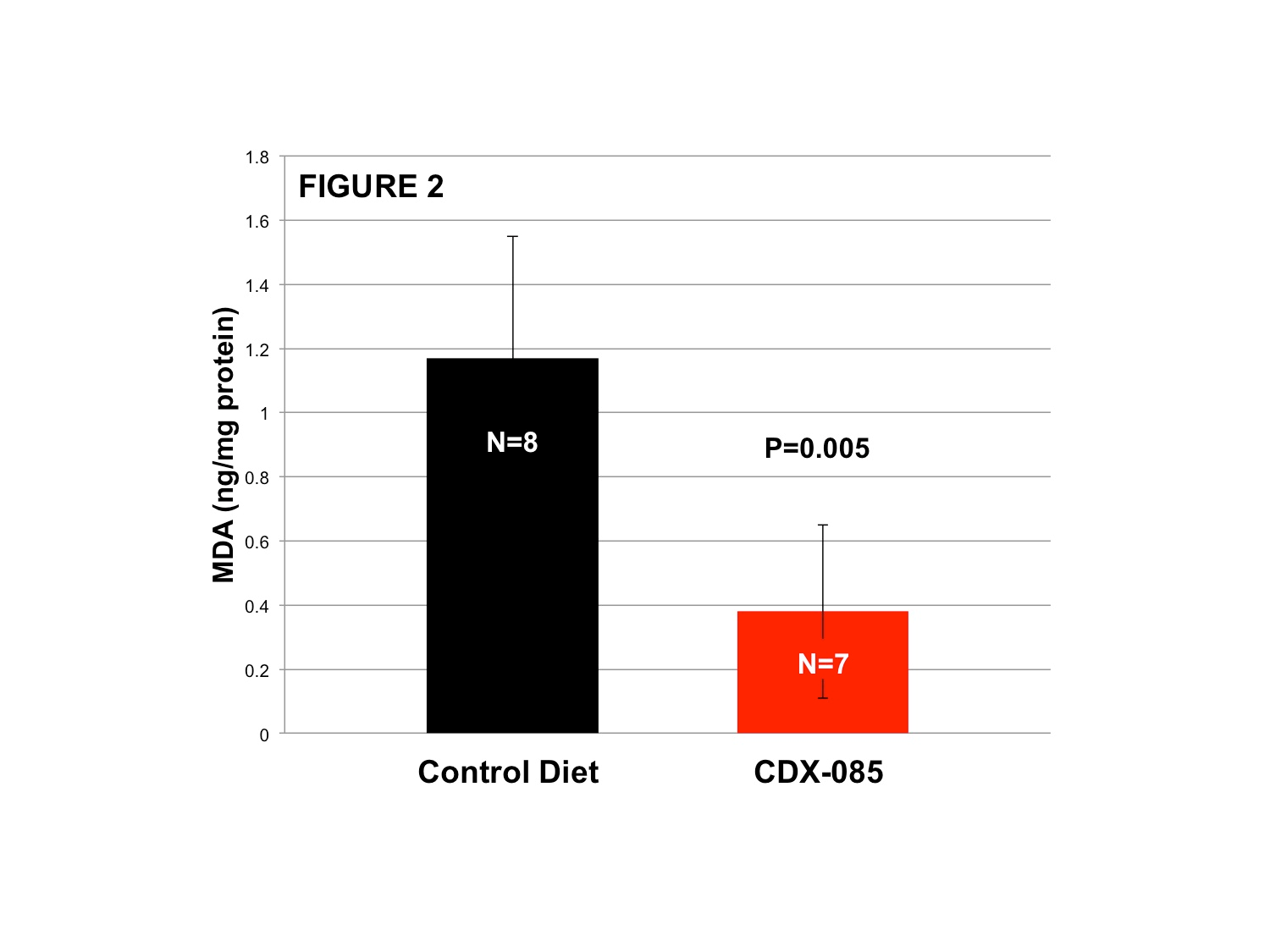
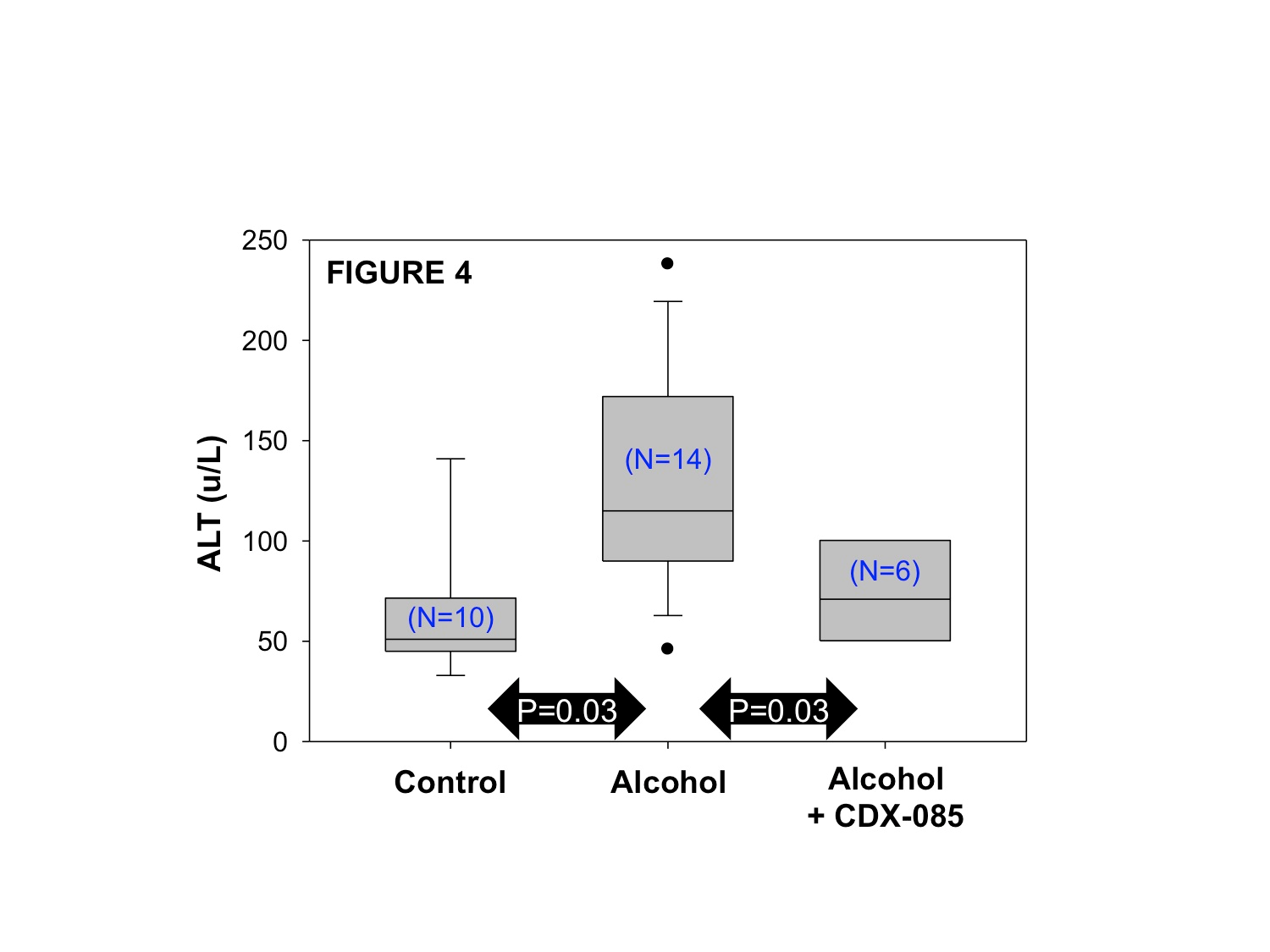
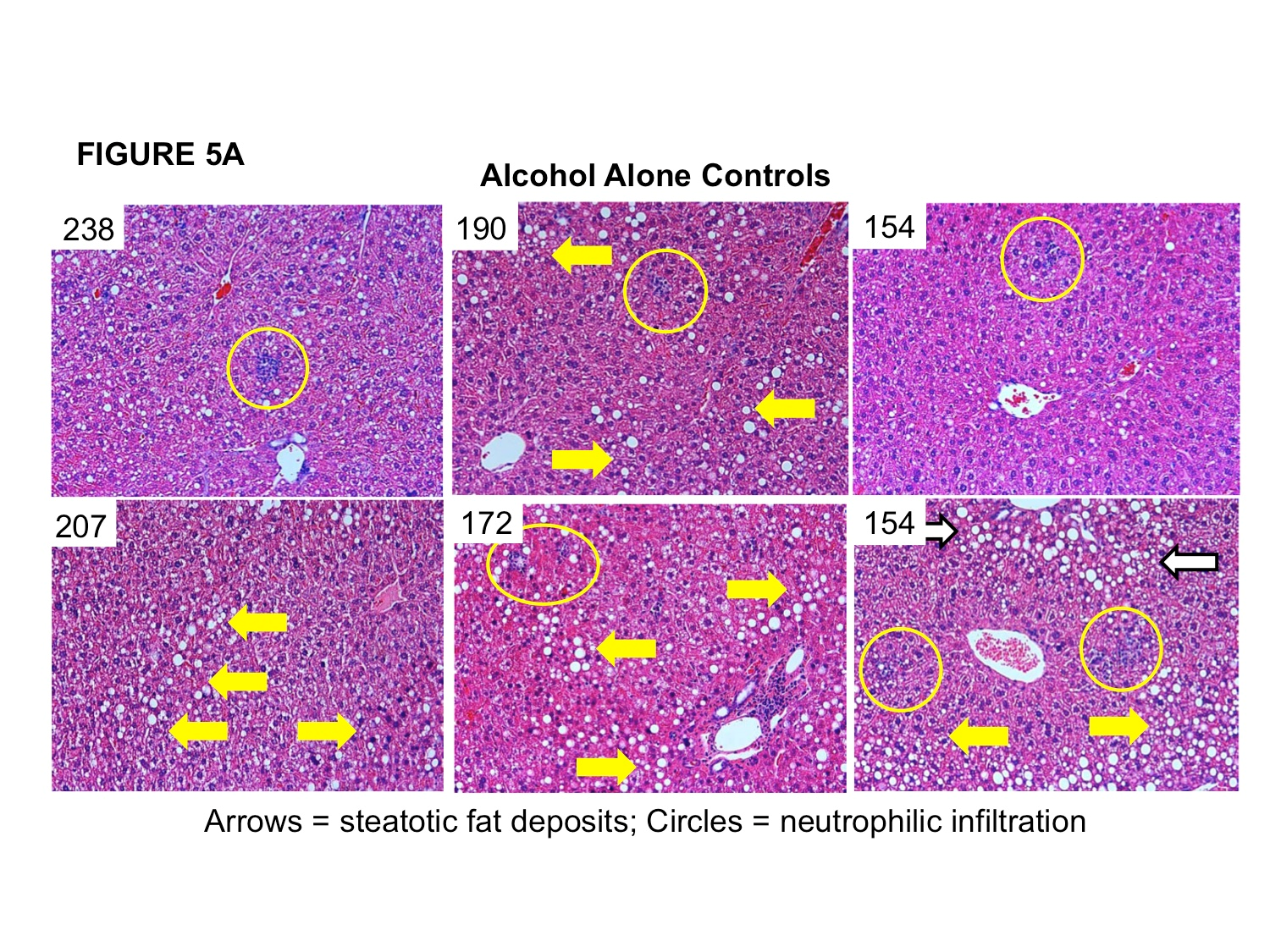
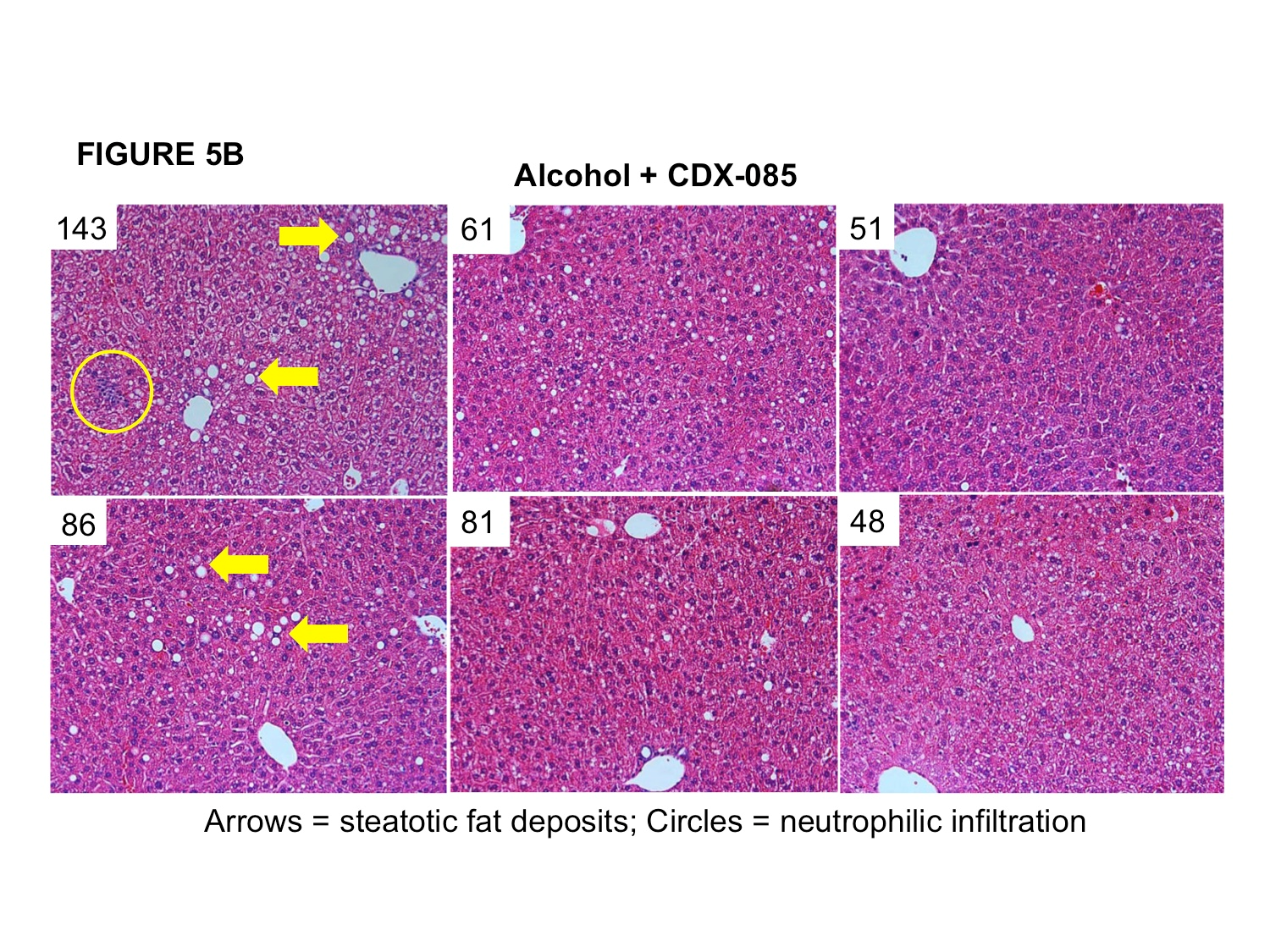
In unstressed mice, an 11-day oral administration of 500mg/kg of CDX-085 to mice delivered high levels of ASTX to plasma and liver and importantly liver mitochondria. Eight weeks of CDX-085 treatment (500mg/kg in chow) significantly reduced systemic oxidative stress in vivo measured as diminished MDA levels (TBAR assay, p=0.005). To further evaluate ASTX influence on disease states, we investigated the ability of CDX-085 to reduce alcohol-induced liver dysfunction in a published mouse model of increased hepatic oxidative stress, elevated liver enzyme levels (ALT), and histological alterations including macrovesicular steatosis (excessive fat in the liver) and pan-lobular inflammation with infiltrating neutrophil cells leading to frank necrosis. In response to alcohol treatment (ethanol), ALT levels were significantly elevated in the alcohol only group (p=0.03 compared to no alcohol control group) but were significantly reduced to near control levels in mice treated with CDX-085+alcohol (p=0.03 compared to alcohol only group), with no statistically significant difference when compared to the untreated/no alcohol control. Qualitative analysis of hepatic histological samples revealed decreased steatosis (lipid-filled vesicularization) and diminished inflammatory cell infiltration in CDX-085+alcohol treated mice compared to the alcohol only group.
These animal model results support the potential role of CDX-085/ASTX in reducing alcohol-induced liver stress by decreasing pathologically elevated liver enzymes (ALT), reducing histological evidence of disease including steatosis, and reducing inflammatory cell infiltration. This newly released data joins many previously published studies in underscoring the potential for ASTX to ameliorate liver-associated oxidative stress, inflammation, and the resulting pathology of liver disease in addition to aspects of dyslipidemia (hypertriglyceridemia). In support, a recent study in humans showed ASTX administration to biopsy-determined NASH patients significantly reduced both NAFLD Activity Scores (NAS) and steatosis.
INTRODUCTION
The role of oxidative stress and inflammation in liver injury and liver dysfunction as well as the potential for antioxidants like astaxanthin (ASTX) to reduce liver diseases such as NAFLD (non-alcoholic fatty liver disease) and its necro-inflammatory progression NASH (non-alcoholic steatohepatitis) has been established in both animal models and human trials.1-3 Studies utilizing rodent models of liver disease have also demonstrated the efficacy of ASTX in ameliorating liver injury.3-7 ASTX is a safe, orally bioavailable, naturally occurring molecule with strong anti-inflammatory and antioxidant activity.8,9 Normally found in yeast, algae, krill, crustaceans, and salmon, ASTX is routinely added to animal feed to improve health and reproductive capacity. Following oral administration and intestinal uptake, ASTX is shuttled to the liver via chylomicrons with subsequent systemic distribution to many tissues throughout the body (heart, liver, intestine, adipose, lung, kidney) via plasma lipoprotein particles including VLDL, HDL, and LDL. Once in the cell, ASTX accumulates within the lipid structures of various organelles including plasma, nuclear, endoplasmic reticular, and importantly, mitochondrial membranes. Due to its chemical structure, ASTX completely traverses the lipid bilayer component of cell membranes, facilitating its biphasic (aqueous and lipid) anti-oxidant functions. As mitochondria importation of molecules is highly restricted by the cell, mitochondrial ASTX localization allows for the unique regulation of oxidative and nitrosative stress at the source. Mitochondria are also critical to normal metabolic function and often at the heart of cellular decisions including death, metabolic regulation, and aging. In addition to mitochondrial influence, ASTX has the capacity to influence both intracellular inflammatory (NF-kB, etc.) and metabolic pathways directly modulated by oxidative stress mediators (PI3K-AKT, etc.).
ASTX HUMAN EFFICACY AND ANIMAL PROOF OF CONCEPT
Importantly, ASTX was shown in a clinical trial to reduce liver histopathology (steatosis) and disease scoring (NAS) in biopsy-defined NASH patients.3 Additionally, ASTX has been shown to significantly lower important inflammatory and metabolic disease measures including tumor necrosis factor-alpha (TNF-a), low density lipoprotein cholesterol (LDL-C), apolipoprotein B (ApoB), and triglycerides while significantly increasing adiponectin and high density lipoprotein cholesterol (HDL-C) levels.10-12 ASTX has also positively affected markers of oxidative stress in humans including significantly lowering isoprostanes and malondialdehyde (MDA) levels and significantly increasing total antioxidant capacity (TAC) and superoxide dismutase (SOD).12,13
Evaluation of ASTX function in several animal models of diabetes and metabolic dysfunction supports a positive influence of ASTX on blood glucose levels, insulin levels, homeostatic index of IR (measured as HOMA-IR), and adiponectin levels.14-18 One study demonstrated ASTX efficacy nearly equivalent to pioglitazone’s influence on insulin resistance reduction (HOMA-IR).19 ASTX treatment also improved insulin pathway activation and increased critical translocation of the insulin-regulated glucose transporter GLUT4 both in vivo (skeletal muscle) and in cell culture.15,20 Significant improvement in the profiles of hepatic glucose catabolic and regulatory enzymes was also seen in addition to increased activation of hepatic insulin signaling proteins including IRS-1/2, PI3K, and AKT. 15,20 Induced with a high-fat high-fructose diet, the activated stress response, as well as pro-inflammatory signaling pathways JNK-1 and ERK-1, were significantly reduced in ASTX treated groups. 18 All these studies clearly demonstrate a robust capacity of ASTX to support many important aspects of insulin functionality in a resistant environment by improving insulin signaling (IRS, AKT, PI3K) and glucose metabolism (enzymes and GLUT4) while protecting these pathways from increased oxidative stress insults evidenced by improved insulin resistance (HOMA, QUICK1) and improved antioxidant status (antioxidant levels, decreased ERK and JNK activation).
The importance of mitochondrial function/dysfunction in metabolic and liver diseases is paramount. Many studies support the strong influence of ASTX on mitochondrial functionality as well as inflammatory and metabolic intracellular signaling in animal and cell-based models.8,9,15,21-24 Specifically, ASTX has been shown to: 1) decrease mitochondrial oxidative stress, 2) stabilize mitochondrial membranes leading to decreased pro-apoptotic mediators (Bax, cytochrome C release) and increased anti-apoptotic mediators (Bcl-2), 3) decrease subsequent activation of JNK pathways that induce cell death, and 4) upregulate PGC-1a, a master regulator of mitochondrial biogenesis, as well as critical metabolic regulators such as CPT-1.21-26
Cardax has previously demonstrated efficacy of the proprietary ASTX prodrug CDX-085 in significantly reducing lipids, atherogenesis, and thrombosis in mouse models of disease.27,28 Earlier generation Cardax ASTX prodrugs have also been shown to ameliorate thrombosis and platelet activation as well as decrease inflammation in animal models.29,30
STUDY DETAILS
Here, in collaboration with Dr. Steven Weinman, M.D., Ph.D. (Director of the Liver Center at University of Kansas, Kansas City, Kansas), Cardax evaluated the capacity for a proprietary ASTX prodrug (CDX-085) to ameliorate oxidative stress-induced hepatic dysfunction in an animal model of disease. Initially, 11-day oral administration of 500 mg/kg of CDX-085 to mice delivered high levels of ASTX to plasma and liver and, importantly, liver mitochondria (Figure 1) in contrast to control mice that exhibited no detectable ASTX. Calculations based on differential centrifugation techniques support approximately 4% of liver ASTX content was localized to liver mitochondrial fractions.
Next, the ability of orally administered CDX-085 to decrease systemic oxidative stress was evaluated in mice using measures of the lipid oxidation product malondialdehyde (MDA, TBARS assay). Eight weeks of CDX-085 treatment (500 mg/kg in chow, N=8 control, N=7 +CDX-085) significantly reduced MDA levels demonstrating the ability of CDX-085/ASTX to reduce systemic oxidative stress in vivo (Figure 2, p=0.005, two-sided T-test).
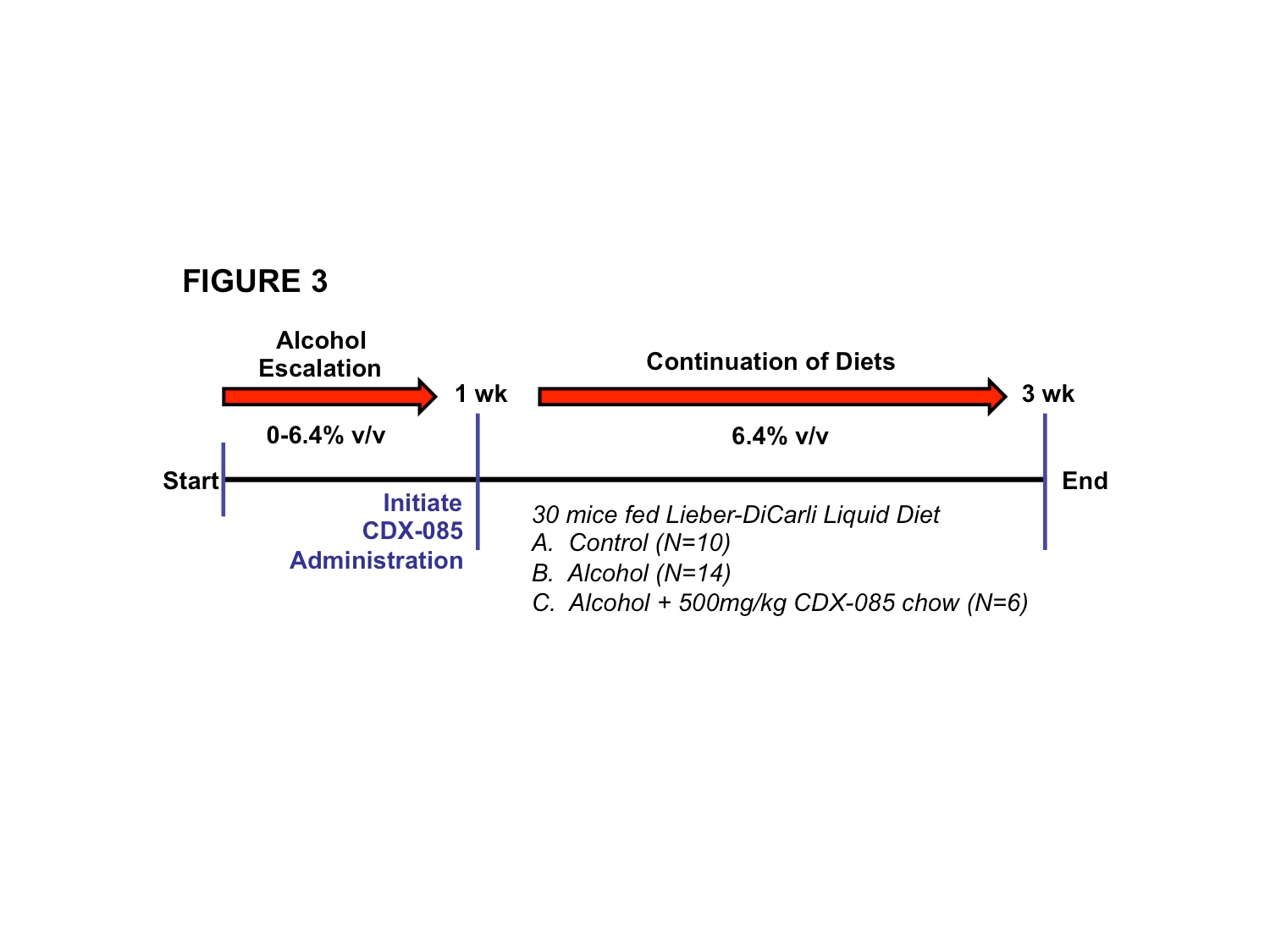
To evaluate CDX-085 effects in a disease model, we evaluated the ability of CDX-085 to reduce alcohol-induced liver dysfunction in a mouse model expressing Hepatitis C Virus structural proteins (core, E1, E2, p7) in a background of diminished mitochondrial antioxidant capacity (lacking one copy of manganese superoxide dismutase, MnSOD).31 When exposed to high levels of dietary alcohol (ethanol), these genetic alterations predispose mice to liver stress exhibited as elevated liver enzyme levels (ALT) and histological alterations including macrovesicular steatosis (excessive fat in the liver) and pan-lobular inflammation with infiltrating neutrophil cells leading to frank necrosis.
Following alcohol escalation for one week (escalating 0-6.4% v/v ethanol), CDX‑085 was administered at 500mg/kg in Lieber-DiCarli liquid diet +/- alcohol for 2 additional weeks (Figure 3). A total of 30 mice were evaluated for ALT levels and histological liver alterations (N=10, control; N=14, alcohol alone; N=6 alcohol+CDX-085). As shown in Figure 4, ALT levels were significantly elevated in alcohol treated mice (p=0.03). This pathological elevation was significantly reduced to near normal levels in mice treated with CDX-085+alcohol (Figure 4, p=0.03 compared to alcohol alone, no statistically significant difference when compared to control alone).
In response to alcohol-stress, mice in this model develop dramatic steatosis (intrahepatocellular fat deposits) and demonstrate neutrophilic infiltration of the liver (Figure 5A, corresponding ALT levels for individual mice in top left corners of micrograph). Qualitative analysis of hepatic histological samples revealed decreased steatosis (lipid-filled vesicularization) and diminished inflammatory cell infiltration in CDX-085+alcohol treated mice compared to alcohol alone mice (Figure 5B, corresponding ALT levels for individual mice in top left corners of micrograph).
In support of these findings, ASTX has previously been shown to significantly: 1) reduce pathologically elevated liver enzyme levels (ALT and AST), 2) decrease histological hepatocytic necrosis, liver lobule damage, and pericellular bridging fibrosis, and 3) attenuate liver fibrosis in mouse models of surgically induced (bile duct ligation) and chemically induced liver injury (carbon tetrachloride).32 Additionally, in an animal model more reflective of the environment-induced hepatic dysfunction seen in human clinical settings, ASTX significantly decreased plasma triacylglycerides and reduced liver TAGs albeit not significantly.33 Several published studies have likewise shown the capacity of ASTX to reduce plasma and liver triglyceride levels in animal models of metabolic dysfunction.7,14,17,28
In summary, these in vivo results support the potential role of CDX-085/ASTX in reducing alcohol-induced liver stress by decreasing pathologically elevated liver enzymes (ALT) and reducing histological evidence of disease including steatosis and inflammatory cell infiltration. Importantly, a clinical trial demonstrated the ability of ASTX to significantly reduce liver histopathology (steatosis) and disease scoring (NAS) in biopsy-defined NASH patients3. Data presented here joins many previously published studies in underscoring the potential for ASTX to ameliorate liver-associated oxidative stress, inflammation, and the resulting pathology of liver disease in addition to aspects of dyslipidemia (hypertriglyceridemia).
ACKNOWLEDGEMENT
This work was supported in part by a grant from the National Institute on Alcohol Abuse and Alcoholism (AA018922).
References
- Pacana, T. and Sanyal, A.J., Curr. Opin. Clin. Nutr. Metabol. Care, 15(6):641-648, 2012.
- Sanyal, A.J. et al., New Eng. J. Med., 362:1675-1685, 2010.
- Ni, Y. et al., Science Reports 5:17192, 2015.
- Kang, J.-O., et al. Meth. Findings Exp. Clin. Pharm., 23(2):79-84, 2001.
- Curek, G.D., Cort, A., Ycel, G. et al., Toxicology, 267(1-3):147-153, 2010.
- Shen, M., Chen, K., Lu, J., Cheng, P., Xu, et al., Med. Inflam., Vol. 2014, Art. ID 954502, 2014.
- Yang, Y., Seo, J.M., Nguyen, A., et al., J. Nutr., 141: 1611-1617, 2011.
- Ambati, R.R. et al., Marine Drugs 12:128-152, 2014.
- Kishimoto, Y. et al., Eur. J. Nutr. on-line 14(35), 2016.
- Uchiyama, A. and Okada, Y., J. Clin. Biochem. Nutr., 43 Suppl (1);38-43, 2008.
- Yoshida, H. et al., Atherosclerosis 209(2):520-523, 2010.
- Choi, H.D. et al., Plant Foods Hum. Nutr. 66(4):363-369, 2011.
- Choi, H.D. et al., Phytotherapy Res. 25(12):1813-1818, 2011.
- Bhuvaneswari, S. et al., Process Biochem. 45:1406-1414, 2010.
- Arunkumar, E. et al., Food & Function 3(2):120-126, 2011.
- Aoi, W. et al., Biochem. Biophys. Res. Comm. 366:892-897 2008.
- Hussein, G. et al., Life Sci. 80:522-529, 2007.
- Bhuvaneswari, S. et al., Can. J. Physiol. Pharmacol. 90:1544-1552, 2012.
- Preuss, H.G. et al., Int. J. Med. Sci. 8(2):126-138, 2011.
- Ishiki, M. et al., Endocrinol. 154(8):2600-2612, 2013.
- Park, J.S. et al., J. Anim. Sci. 91:268-275, 2013.
- Wolf, A.M. et al., J. Nutr. Biochem. 21(5):381-389, 2010.
- Lee, D.-H. et al., Food Chem. Toxicol. 49(1):271-280, 2011.
- Chan, K.-C. et al., J. Food Sci. 77(2)H76-H80, 2012.
- Liu, X. et al., Brain Res. 1254:18-27, 2009.
- Liu, P.-H. et al., J. Clin. Biochem. Nutr. 54(2):86-89, 2014.
- Khan, S.K. et al., Thromb. Res. 126:299-305, 2010..
- Ryu, S.K. et al., Atherosclerosis 222(1):99-105, 2012.
- Lockwood, S.F. and Gross, G.J., Cardiovasc. Drug Rev. 23(3):199-216, 2005.
- Lauver, D.A. et al., Pharmacol. 82(1):67-73, 2008.]
- Tumurbaatar, B. et al., Am. J. Pathol. 183(6):1803-1814, 2013.
- Ye, J. Front. Med. 7(1):14-24, 2013.
- Yasukawa, T. et al., J. Biol. Chem. 280(9):7511-7518, 2005.